Acute Oxalate Nephropathy
- Acute oxalate nephropathy is a rare cause of acute kidney injury (AKI).
- Acute oxalate nephropathy can be primary (type 1, type 2 and type 3 hyperoxaluria) or secondary.
- The primary hyperoxalurias are rare genetic diseases caused by deficiencies in glyoxylate metabolism
- Type 1 hyperoxaluria: the most common (70–80%) of the hereditary defects. There is deficiency of hepatic microsomal alanine glyoxylate aminotransferase due to mutation in the AGXT gene (expressed only in the liver). Consequently, glyoxylate is not adequately metabolized to glycine, which results in the overproduction of oxalate and glycolate. The clinical expression of type I is hyperoxaluria quite heterogeneous.
- Type 2 hyperoxaluria: second most common of the hereditary defects. There is deficiency of glyoxylate reductase/hydroxypyruvate reductase enzyme due to mutation in the GRHPR gene (widespread tissue expression). Consequently, there is increased oxalate and L-glyceric acid, usually with less severe kidney disease than in type 1. It is believed that many more patients with this form of the disease may go undiagnosed because of the less severe clinical course and the fact that the disease is not widely recognized.
- Type 3 hyperoxaluria: rare form of hereditary oxalosis. There is deficiency of mitochondrial 4-hydroxy-2-oxoglutarate aldolase enzyme due to mutation in the OAT gene (widespread tissue expression) with resultant aberrant hydroxyproline metabolism and increased oxalate. These patients usually do not progress to ESRD. It is speculated that patients with primary hyperoxaluria type III express a protective urinary, environmental, epigenetic, or genetic factor that helps to decrease urinary oversaturation for calcium oxalate and therefore the risk of recurrent stone disease or progressive nephrocalcinosis.
- Secondary hyperoxaluria: can occur due to increased dietary oxalate intake (Averrhoa carambola, vitamin C, beets, chocolate, peanuts, black tea, spinach, rhubarb, Chaga mushroom, piridoxylate, ethylene glycol, etc.), increased absorption of oxalate from the bowel (Crohn’s disease, celiac disease, chronic pancreatitis, small bowel resection, diabetic gastroenteropathy, pancreatic adenocarcinoma, systemic sclerosis, Roux-en-Y bypass, hemicolectomy, gastric bypass, jejuno-ileal bypass, bariatric surgery, cystic fibrosis, orlistat, octreotide), increased production/decreased degradation of oxalate (Absence of Oxalobacter formigenes colonization) and increased colonic permeability to oxalate (Clostridium difficile colitis).
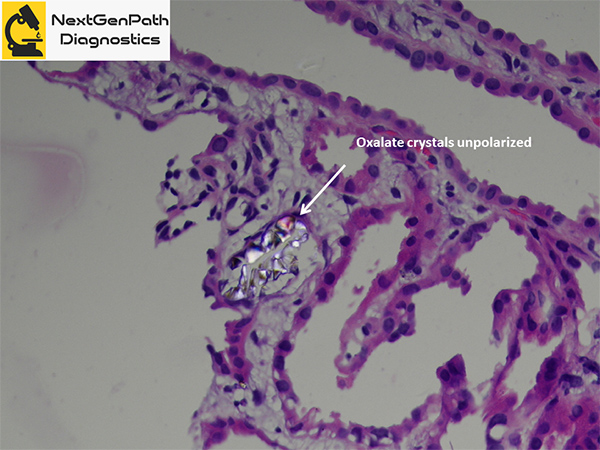
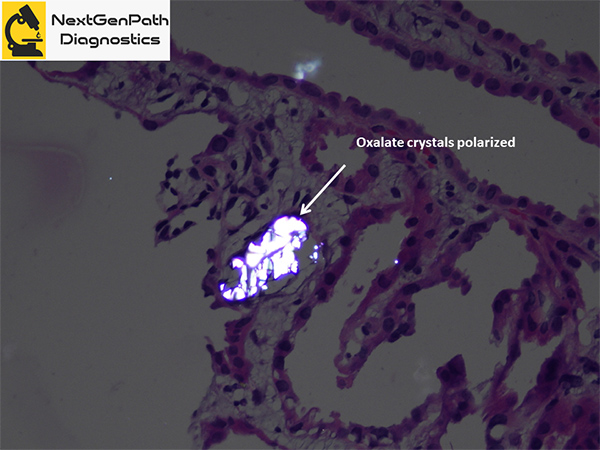
- Renal biopsy: acute tubular injury, with frequent intratubular crystals, which are clear on hematoxylin and eosin stain under light microscopy and show multicolored birefringence under polarized light, with classic fan-shaped morphology.
- Treatment: Only liver transplantation is able to cure type I primary hyperoxaluria and therefore pre-emptive liver or sequential or combined liver and kidney transplantation are the methods of choice in patients with this form of the disease. Treatment of secondary hyperoxaluria includes hydration and dietary interventions, including the adoption of a normal calcium, low-oxalate diet. In addition, the use of diuretics for fluid overload, alkali therapy to alkalinize the urine and pyridoxine to covert glyoxalate (the main precursor of oxalate) to glycine have been tried with variable success.
References
- Hoppe B. An update on primary hyperoxaluria. Nat Rev Nephrol. 2012;8:467-75.
- Lumlertgul N, Siribamrungwong M, Jaber BL, Susantitaphong P. Secondary Oxalate Nephropathy: A Systematic Review. Kidney Int Rep. 2018;3:1363-1372.
- Fogo AB, Lusco MA, Najafian B, Alpers CE. AJKD Atlas of Renal Pathology: Oxalosis. Am J Kidney Dis. 2017;69:e13-e14.