Wilson disease (WD) is an inherited disorder of copper metabolism, caused by mutations in ATP7B, which encodes a copper transporting protein characterized by pathological copper accumulation in different organs.
The commonly affected organs include liver, brain, eyes, red blood cells and cartilage.
WD affects 1:30,000 to 1:100,000 individuals. There are some unique populations where there is an increased disease incidence thought to be related to consanguinity, with presence of often dominant mutations of ATP7B.
The clinical signs and symptoms of WD are diverse. Many patients present with signs of liver disease, such as fatty liver, elevated serum biomarkers of liver injury or inflammation, or decompensated (symptomatic) cirrhosis.
Neurological manifestations are also common; patients often present with a movement disorder with or without dysarthria (unclear articulation of speech), gait and posture disturbances, drooling, and dysphagia (difficulties during any phase of swallowing).
Patients may also have personality and mood disorders or even psychosis.
Pathological copper accumulation in the eyes can cause characteristic Kayser-Fleischer rings (golden, brown or green colouration at the periphery of the cornea).
To establish the diagnosis, a combination of clinical features and laboratory parameters are required. Several laboratory tests can measure copper or ceruloplasmin (the major copper carrying protein in the blood) levels in the serum, liver or urine. Moreover, DNA sequencing can be used to identify disease causing mutations.
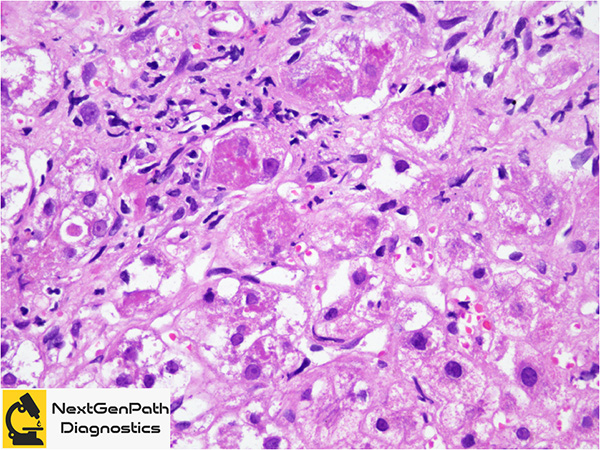
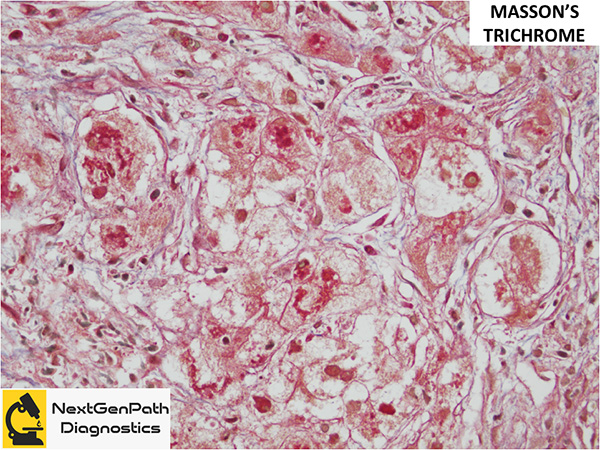
Role of liver biopsy in WD: The biopsy may be done at different points in the investigation of liver disease, before or after the suspicion for WD arose clinically, depending on patient presentation, degree of suspicion and as clinical findings/events unfold. Clinical findings that should prompt liver biopsy for histology and copper quantitation include: 1. KF rings present, ceruloplasmin >20 mg/dL, 24-h urine copper >40 μg; 2. KF rings absent, ceruloplasmin <20 mg/dL, 24-h urine copper <40 μg; KF rings absent, ceruloplasmin <20 mg/dL, 24-h urine copper >40 μg.
Management: WD can be successfully managed if the disease is diagnosed before serious liver or brain damage occurs. Treatment for WD is based on therapeutics that correct copper homeostasis: copper chelators (d-penicillamine and trientine) that increase urinary copper excretion and zinc salts that decrease copper absorption from the gut. In patients with acute liver failure or decompensated cirrhosis, liver transplantation is indicated, which phenotypically corrects the mutation in ATP7B, therefore restoring copper homeostasis.
References
Guindi M. Wilson disease. Semin Diagn Pathol. 2019;36:415-422.
Członkowska A, Litwin T, Dusek P, et al. Wilson disease. Nat Rev Dis Primers. 2018;4:21.