Lipid storage diseases affecting skeletal muscle are rare inborn errors of metabolism characterized by progressive
muscle weakness, exercise intolerance, exercise-induced muscle stiffness, cramps and
recurrent episodes of rhabdomyolysis.
All the lipid storage myopathies (LSM) have an autosomal recessive mode of inheritance.
There are mainly four types LSMs: primary carnitine deficiency (PCD), multiple acyl-coenzyme A dehydrogenase
deficiency (MADD), neutral lipid storage disease with ichthyosis, and neutral lipid storage disease with myopathy.
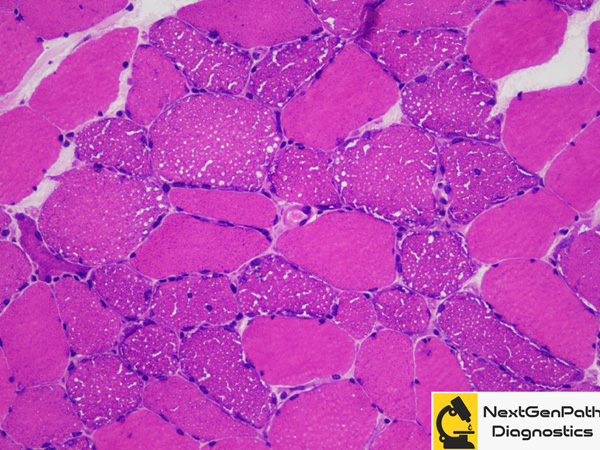
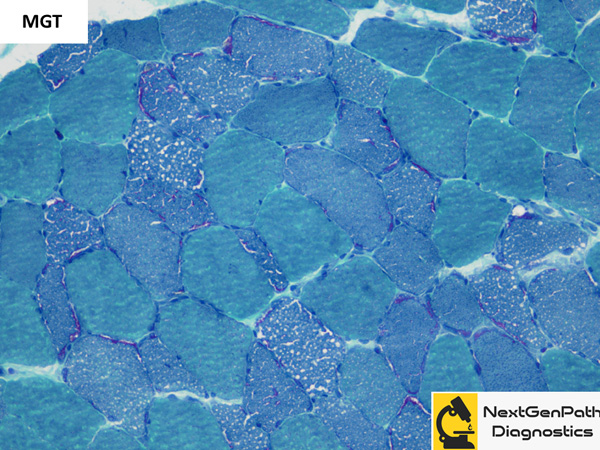
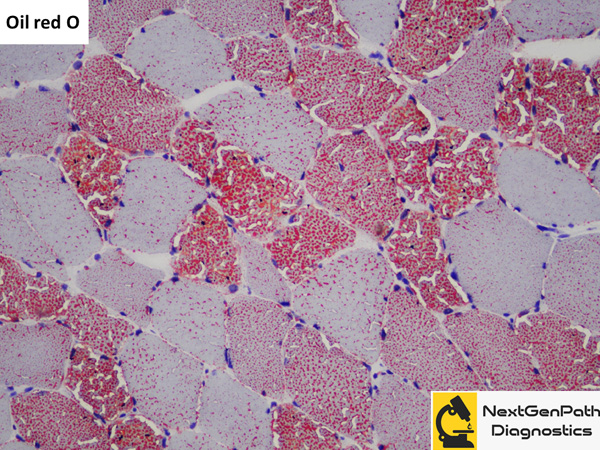
Biopsy: In a normal muscle, the proportion of fat globules in type 1 fibers is more than that of type
2 fibers. The fat globules also appear slightly larger in type 1 fibers. In lipid storage myopathies, the lipid droplets
exceed that of normal giving the sarcoplasm a vacuolated appearance on H&E stain which gives positive reactions
for lipid stains such as Oil red O and Sudan black B.
Making an accurate diagnosis, by specific laboratory tests is important for LSMs as some of the patients are
treatable: individuals with PCD show dramatic improvement with high-dose oral L-carnitine supplementation
and increasing evidence indicates that MADD due to ETFDH mutations is riboflavin responsive.
References
Gaspar BL, Vasishta RK, Radotra BD. Myopathology: A Practical Clinico-Pathological Approach to
Skeletal Muscle Biopsies. Springer Nature: Singapore, 2019.